
Halogen bonds in crystal TTF derivatives: an ab initio quantum mechanical study
Abstract
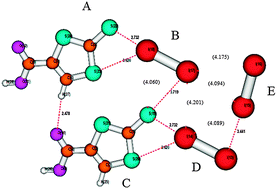
The stabilisation energies of five ionic and neutral organic crystal structures containing various halogen bonds (I⋯I, Br⋯Br, I⋯Br, I⋯S and Br⋯S) were calculated using the DFT-D3 method (B97D/def2-QZVP). Besides them, the ionic I3−⋯I2 and neutral I2⋯I2, complexes (in the crystal geometries) were also studied. The nature of the bonds was deduced from the electrostatic potential evaluated for all subsystems. In almost all the cases, the σ-hole was positive; it was negative only for the ionic I3− system (although more positive than the respective belt value). The strongest halogen bonds were those that involved iodine as a halogen-bond donor and acceptor. Among ionic X⋯I3− and neutral X⋯I2 and X⋯Y dimers, the neutral X⋯I2 complexes were, surprisingly enough, the most stable; the highest stabilisation energy of 13.8 kcal mol−1 was found for the I2⋯1,3-dithiole-2-thione-4-carboxylic acid complex. The stabilisation energies of the ionic I3−⋯I2 and neutral I2⋯1,3-dithiole-2-thione-4-carboxylic acid (20.2 and 20.42 kcal mol−1, respectively) complexes are very high, which is explained by the favourable geometrical arrangement, allowing the formation of a strong halogen bond. An I⋯I halogen bond also exists in the neutral I2⋯I2 complex, having only moderate stabilisation energy (3.9 kcal mol−1). This stabilisation energy was, however, shown to be close to that in the optimal gas-phase L-shaped I2⋯I2 complex. In all the cases, the dispersion energy is important and comparable to electrostatic energy. Only in strong halogen bonds (e.g. I3−⋯I2), the electrostatic energy becomes dominant.
 Please wait while we load your content...
Please wait while we load your content...

