
Improved accuracy for label-free absolute quantification of proteome by combining the absolute protein expression profiling algorithm and summed tandem mass spectrometric total ion current†
Abstract
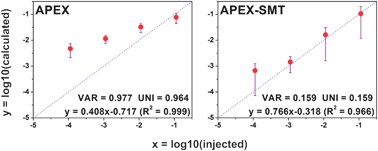
Proteome scale absolute quantification is fundamental for the quantitative understanding of an organism. The unsatisfactory accuracy for protein abundance estimation of current algorithms has been partially improved by the Absolute Protein EXpression profiling (APEX) algorithm, which implements the prior expectations of peptides' appearances in the calculation of protein abundances. However, the abundance feature (AF) in APEX is the spectral count (SC); an AF suffers from a narrow dynamic range, thus, unsatisfactory accuracy. Therefore, we adopted another tandem mass spectrometric (MS/MS) level AF called Summed MS/MS Total ion current (SMT), which cumulates the MS/MS fragment intensities rather than simply counting the MS/MS spectra, to surmount this particular deficiency. The combination of APEX and SMT (abbreviated as APEX-SMT) is capable of improving the accuracy of absolute quantification by reducing the average relative deviation by ∼55–85% compared to that of APEX, through a series of tests on the Universal Proteomics Standard sample with a dynamic range of 5 orders of magnitude (UPS2). The algorithm could also be used for relative quantification. When applied to the relative quantification of a publicly available benchmark dataset, APEX-SMT could provide comparable accuracy to APEX. All these results suggest that APEX-SMT is a promising alternative to APEX for proteome quantification.
 Please wait while we load your content...
Please wait while we load your content...

