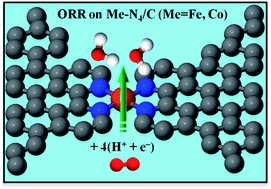
In this study, we performed first-principles density functional theory (DFT) calculations to investigate the reaction pathway of oxygen reduction reaction (ORR) on Me–N4 (Me = Fe, Co, or Ni) catalytic clusters formed between pores in graphene supports. Our DFT results indicate that formation of Me–N4 clusters at the edges of graphitic pores is energetically feasible. The catalytic mechanism of the Me–N4 clusters for ORR was elucidated by calculating the binding energies of ORR intermediates O2, O, OH, OOH, HOOH, and H2O on these clusters. It was found that O2 could be chemisorbed on the Co–N4 and Fe–N4 clusters but not on the Ni–N4 clusters. Thus, the Co–N4 and Fe–N4 clusters are predicted to be active for catalyzing ORR. Moreover, we observed that the O–O bond in HOOH was broken in the lowest-energy adsorption configuration of HOOH on the Fe–N4 and Co–N4 clusters. This process of breaking the O–O bond suggests that the Fe–N4 and Co–N4 clusters between graphitic pores could promote 4e− reduction of O2 with a single active site. Here, our theoretical study provides an explanation to the experimental findings of 4e− ORR on nitrogen-derived transition metal/carbon electrocatalysts. Furthermore, we illustrated that the ORR activity of the Me–N4 clusters could be gauged by an electronic descriptor related to the d-electron orbitals of the transition metal atom.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


