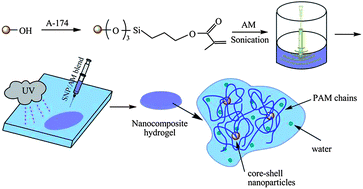
The poor mechanical strength of traditional synthesized hydrogels remains the greatest challenge to their performance as tissue engineering materials. Unique combinations of hard and soft components were used to design and synthesize elastomeric nanocomposite hydrogels with a core–shell microstructure. The nanocomposite hydrogels consisting of multifunctional cross-links of silica nanoparticles (SNPs) as the nucleus and poly(acrylamide) (PAM) as the shell were prepared by mixing both components in solution, and photo-crosslinking by UV radiation. Transmission electron microscopy (TEM) observations confirmed the existence of a core–shell morphology, and dynamic light scatting (DLS) measurements were applied to monitor the particle conformation changes near the critical gelation condition. The results indicated that the concentration of the core significantly affected the mechanical properties of the hydrogel and the unique three-stage arrangement process exhibited an average effective functionality around 28. The molecular weight of grafted polymer chains was monitored and it was found that the grafted PAM chain had a high value of 2.46 × 105 g mol−1, and it remained almost constant when the SNP concentration (CSNP) exceeded the critical value. The mechanical behavior of the nanocomposite hydrogels was studied with elongation under large strains and it was found that the hydrogels exhibited excellent tensile properties, including a modulus 9–38 kPa, a fracture strain 700–1000%, and a fracture stress 80–250 kPa, depending on the SNP concentrations. Stress relaxation experiments indicated that the nanocomposite hydrogels were more viscous than comparable chemical gels and were more efficient at the energy dissipation process, which is related to the orientation of SNPs and relaxation of the PAM chains. Further development of the synthetic platform would potentially lead to valuable knowledge for the design of high performance hydrogels in biomedical and pharmaceutical applications.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


