
Dynamics of the F− + CH3Cl → Cl− + CH3F SN2 reaction on a chemically accurate potential energy surface
Abstract
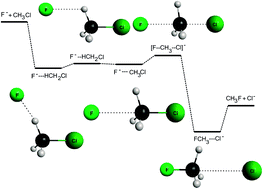
Though bimolecular nucleophilic substitution (SN2) reactions play a fundamental role in chemistry, chemically accurate full-dimensional global analytical potential energy surfaces (PESs) have not been developed for these systems. These PESs govern the motion of the atoms in a chemical reaction; thus, the knowledge of the PES is essential to study the dynamics and atomic-level mechanisms. Here, we report a full-dimensional ab initio PES for the F− + CH3Cl → Cl− + CH3F reaction, the PES has an estimated average accuracy of about 0.5 kcal mol−1. Quasiclassical trajectories on this PES reveal that the direct rebound mechanism dominates at high collision energies, whereas the reaction is mainly indirect at low collision energies, where the formation of long-lived hydrogen-bonded and C3v ion–dipole entrance-channel complexes play a major role in the dynamics. A direct stripping mechanism is also found at large impact parameters resulting in significant forward scattering, whereas the direct rebound mechanism scatters towards backward directions. At high collision energies the reaction can be controlled by orienting the reactants into a reactive F− + H3CCl orientation, whereas at low collision energies the initial orientation is not always maintained, because the long-range ion–dipole interactions efficiently steer the reactants into a reactive orientation even if F− initially approaches the non-reactive side of CH3Cl. Mode-specific vibrational distributions show that the reaction produces vibrationally hot CH3F molecules with excited CF stretching, especially at low collision energies.
 Please wait while we load your content...
Please wait while we load your content...

