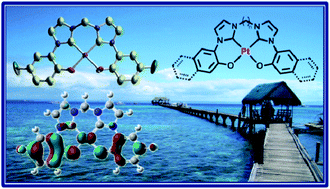
The synthesis, structures and photophysical properties of the charge-neutral Pt(II) complexes (1–6) and their Pd(II) (7) and Ni(II) (8) congeners supported by tetradentate dianionic bis[phenolate-(N-heterocyclic carbene)] ligands are described. The X-ray crystal structures of two solvatomorphs of 2, which has p-F substituents on the tetradentate ligand, have been determined. The photophysical properties of all the complexes were examined. In THF solutions, 1–4 display deep blue phosphorescence (λmax = ∼440–460 nm, Φe = 3–18% and τ = 0.5–3.5 μs). In solutions at room temperature, 5–8 show profoundly different luminescence properties from being virtually non-emissive (Φe < 10−3) for 6–8 to highly emissive (Φe = 15%) with much red-shifted phosphorescence (λmax = ∼530 nm) and a long emission lifetime (τ = 47.2 μs) in the case of 5. Time-dependent density functional theory (TDDFT) calculations reveal that the tetradentate bis(phenolate-NHC) ligands in 1–4 provide a rigid scaffold for preserving a tightly bound Pt(II) in a square-planar coordination geometry in the T1 as in the S0 states and the blue emission is derived from the T1 state having predominant ligand (πAr–O)-to-ligand (π*NHC) charge transfer (LLCT) character. A switch of orbital parentage from LLCT to ligand-centred (LC) π–π* is responsible for the long emission lifetime and vibronically structured emission displayed by 5 when compared to that of 1–4 and 6. Both femtosecond time-resolved fluorescence (fs-TRF) and nanosecond time-resolved emission (ns-TRE) measurements were conducted on 2 and 4 to directly probe the excited-state dynamics after photoexcitation. Excellent thermal stability of the fluorine-free complex 4 and its higher emission quantum yield (relative to 1 and 3), and using 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) as host material, led to the fabrication of highly efficient deep blue OLEDs with peak current efficiency of 24 cd A−1 and white organic light-emitting devices (WOLEDs) with peak current efficiency of 88 cd A−1.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


