
Combined ligand and structure based binding mode analysis of oxidosqualene cyclase inhibitors†
Abstract
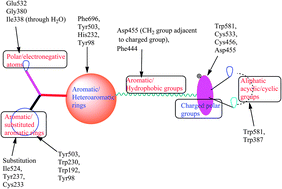
In this present investigation, computational analysis was performed on a data set of OSC inhibitors in order to perform its binding mode analysis and lead identification. The pharmacophore analysis and QSAR studies revealed that the radius of the Aro/Hyd and/or Hyd properties and the polar properties are important for the interaction and activity elicitation. The docking results derived from the analysis also explain that the aromatic amino acids predominantly present in the active site and the polar groups are separated by the aromatic/hydrophobic regions. The significant aromatic amino acids Phe696, Phe444 and His232 make π–π stacking or CH–π interactions with the functional groups (aromatic or aliphatic) present in the inhibitors. The polar groups present in the compounds interact with Asp455 or Trp581 or Trp387 or Ile338 or Gly380 (all through water) or with Glu532. The natural products analyzed on this target provided significant docking scores and its mode of interactions are similar to the OSC inhibitors, which can be used for the treatment of obesity.
 Please wait while we load your content...
Please wait while we load your content...

