
Discovery of N-(4-sulfamoylphenyl)thioureas as Trypanosoma brucei leucyl-tRNA synthetase inhibitors
Abstract
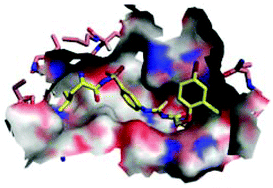
Human African trypanosomiasis (HAT) is one of the most neglected diseases in the tropic regions, which is fatal if not treated in time. There is an urgent need for new therapeutics, especially those in new chemical classes. Leucyl-tRNA synthetase (LeuRS) has been paid much attention as a recently clinically validated antimicrobial target. Our group has previously reported T. brucei LeuRS (TbLeuRS)
- This article is part of the themed collection: In Celebration of Andrew D. Hamilton’s Career in Chemistry
 Please wait while we load your content...
Please wait while we load your content...

