
Multi-edge X-ray absorption spectroscopy of thorium, neptunium and plutonium hexacyanoferrate compounds†
Abstract
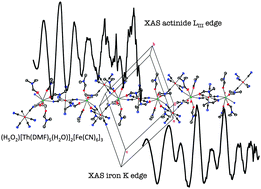
Although transition metal cyano bimetallic compounds have been well known for decades for their interesting optical and magnetic properties, reports on actinide hexacyanoferrate compounds are scarce. For instance, a thorough structural description is still lacking. Another question is the possible covalency or charge transfer effects in these materials that are known to foster electron delocalization with a large variety of transition metal
 Please wait while we load your content...
Please wait while we load your content...

