
A monomeric form of iNOS can rationalise observed SAR for inhibitors of dimerisation: quantum mechanics and docking compared†
Abstract
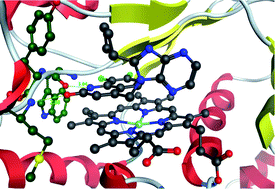
Two series of compounds that inhibit dimerisation of iNOS have been reported in the literature and studied crystallographically. We have applied a range of docking techniques to these compounds and shown that the geometry of the complexes can be reproduced well but that even within a series of related compounds the docking scores do not rank compounds well. Quantum mechanical studies using a model system for the protein and a range of density functionals are able to reproduce the geometry of the complexes and the corresponding affinities. The combination of docking to generate geometries and quantum mechanical calculations to predict complexation energies is a powerful one for rationalising observed changes and designing improved compounds.
 Please wait while we load your content...
Please wait while we load your content...