
Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome†
Abstract
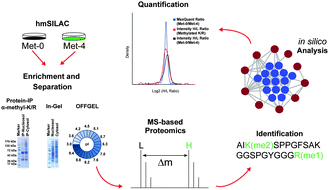
Protein methylation is a post-translational modification (PTM) by which a variable number of methyl groups are transferred to lysine and arginine residues within proteins. Despite increased interest in this modification due to its reversible nature and its emerging role in a diverse set of biological pathways beyond chromatin, global identification of protein methylation has remained an unachieved goal. To characterise sites of lysine and arginine methylation beyond histones, we employed an approach that combines heavy methyl stable isotope labelling by amino acids in cell culture (hmSILAC) with high-resolution mass spectrometry-based proteomics. Through a broad evaluation of immuno-affinity enrichment and the application of two classical protein separation techniques prior to mass spectrometry, to nucleosolic and cytosolic fractions separately, we identified a total of 501 different methylation types, on 397 distinct lysine and arginine sites, present on 139 unique proteins. Our results considerably extend the number of known in vivo methylation sites and indicate their significant presence on several protein complexes involved at all stages of gene expression, from chromatin remodelling and transcription to splicing and translation. In addition, we describe the potential of the hmSILAC approach for accurate relative quantification of methylation levels between distinct functional states.
- This article is part of the themed collection: Molecular BioSystems 2013 Proteomics
 Please wait while we load your content...
Please wait while we load your content...