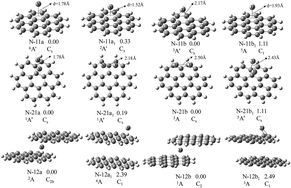
The geometries, electronic and magnetic properties of neutral and negatively charged Mn(coronene)m (M = V and Ti; n, m = 1, 2) complexes were investigated using density functional theory. The results show that one V or Ti atom prefers to occupy the η6-site in M(coronene)0/− complexes and to be sandwiched between the two coronene molecules in M(coronene)20/− complexes. Two metal atoms always form a dimer and interact with one coronene molecule. The calculated vertical electron affinities and transition energies are in good agreement with experimental values. This lends considerable credence to the obtained ground state structure and validates the chosen computational method. The bond formation between metal atom and coronene is accounted for by 3d/4s–π bonds, as revealed by the molecular orbitals plots. The reason why the peripheral ring site binds metal most effectively has been analyzed systematically by π electron content, aromaticity and average charge on carbon atoms. The electron localization function shows that there is perfect electron delocalization in these complexes. Furthermore, the magnetic moments of V(coronene)0/− and Ti(coronene)− are found to be substantially enhanced over the corresponding free metal atom; the magnetic moment of the neutral Ti(coronene) remains unchanged; while the larger size clusters experience a reduction.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


