
Heteroaromaticity approached by charge density investigations and electronic structure calculations†
Abstract
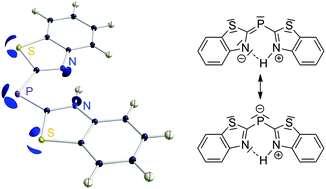
In this paper we present the results of a high-resolution single crystal X-ray diffraction experiment at 15 K on a benzothiazol-substituted phosphane and a subsequent charge density study based on multipole refinement and a topological analysis according to Bader's quantum theory of atoms in molecules. Although two valence shell charge concentrations (VSCCs) in the non-bonding region of each phosphorus and sulfur atom were found, the integration of both heteroatomic basins emphasizes charge depletion. Nevertheless they are attractive for C–H⋯P and C–H⋯S hydrogen bonding in the solid state. The nature of the P–C bonds and the question of aromaticity in the heterocycles were subject to our investigations. The ellipticities along the bonds were analysed to approach delocalization. The source function is employed to visualise atomic contributions to aromaticity. Theoretical calculations have been carried out to compute nuclear chemical shifts, induced ring currents and a variety of delocalization indices. All applied measures for delocalization point in the same direction: while heteroaromaticity is present in the benzothiazolyl substituents, the bridging P–C bonds are only involved marginally, almost preventing total conjugation of the phosphane. The charge density distributions around the phosphorus and the sulfur atoms have very similar features but turn out to be chemically very different from each other. Commonly used simplifying concepts have difficulties in providing a comprehensive view on the electronic situation in the molecule. Our results raise doubts on the validity of the common interpretation of VSCCs as one-to-one representations of Lewis lone pairs.
 Please wait while we load your content...
Please wait while we load your content...

