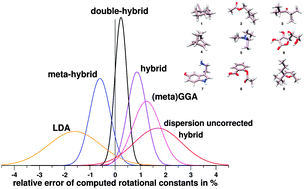
A benchmark set of 25 rotational constants measured in the gas phase for nine molecules (termed ROT25) was compiled from available experimental data. The medium-sized molecules with 18–35 atoms cover common (bio)organic structure motifs including hydrogen bonding and flexible side chains. They were each considered in a single conformation. The experimental B0 values were back-corrected to reference equilibrium rotational constants (Be) by computation of the vibrational corrections ΔBvib. Various density functional theory (DFT) methods and Hartree–Fock with and without dispersion corrections as well as MP2 type methods and semi-empirical quantum chemical approaches are investigated. The ROT25 benchmark tests their ability to describe covalent bond lengths, longer inter-atomic distances, and the relative orientation of functional groups (intramolecular non-covalent interactions). In general, dispersion corrections to DFT and HF increase Be values (shrink molecular size) significantly by about 0.5–1.5% thereby in general improving agreement with the reference data. Regarding DFT methods, the overall accuracy of the optimized structures roughly follows the ‘Jacobs ladder’ classification scheme, i.e., it decreases in the series double-hybrid > (meta)hybrid > (meta)GGA > LDA. With B2PLYP-D3, SCS-MP2, B3LYP-D3/NL, or PW6B95-D3 methods and extended QZVP (def2-TZVP) AO basis sets, Be values, accurate to about 0.3–0.6 (0.5–1)% on average, can be computed routinely. The accuracy of B2PLYP-D3/QZVP with a mean deviation of only 3 MHz and a standard deviation of 0.24% is exceptional and we recommend this method when highly accurate structures are required or for problematic conformer assignments. The correlation effects for three inter-atomic distance regimes (covalent, medium-range, long) and the performance of minimal basis set (semi-empirical) methods are discussed.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


