
Ab initio calculations of the optical absorption spectra of C60-conjugated polymer hybrids
Abstract
A recently developed linear-scaling density-functional theory (LS-DFT) formalism is used to calculate
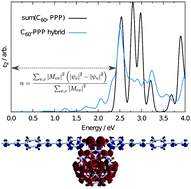
A recently developed linear-scaling density-functional theory (LS-DFT) formalism is used to calculate
L. E. Ratcliff and P. D. Haynes, Phys. Chem. Chem. Phys., 2013, 15, 13024 DOI: 10.1039/C3CP52043A
This article is licensed under a Creative Commons Attribution 3.0 Unported Licence. You can use material from this article in other publications without requesting further permissions from the RSC, provided that the correct acknowledgement is given.
Read more about how to correctly acknowledge RSC content.
 Please wait while we load your content...
Please wait while we load your content...

