Structure and adsorption energy of carbon monoxide molecules adsorbed on the Pt(111) surfaces with various CO coverages in water as well as work function of the whole systems at room temperature of 298 K were studied by means of a hybrid method that combines classical molecular dynamics and density functional theory. We found that when the coverage of CO is around half monolayer, i.e. 50%, there is no obvious peak of the oxygen density profile appearing in the first water layer. This result reveals that, in this case, the external force applied to water molecules from the CO/Pt(111) surface almost vanishes as a result of the competitive adsorption between CO and water molecules on the Pt(111) surface. This coverage is also the critical point of the wetting/non-wetting conditions for the CO/Pt(111) surface. Averaged work function and adsorption energy from current simulations are consistent with those of previous studies, which show that thermal average is required for direct comparisons between theoretical predictions and experimental measurements. Meanwhile, the statistical behaviors of work function and adsorption energy at room temperature have also been calculated. The standard errors of the calculated work function for the water–CO/Pt(111) interfaces are around 0.6 eV at all CO coverages, while the standard error decreases from 1.29 to 0.05 eV as the CO coverage increases from 4% to 100% for the calculated adsorption energy. Moreover, the critical points for these electronic properties are the same as those for the wetting/non-wetting conditions. These findings provide a better understanding about the interfacial structure under specific adsorption conditions, which can have important applications on the structure of electric double layers and therefore offer a useful perspective for the design of the electrochemical catalysts.
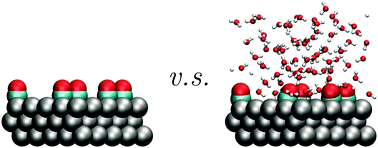
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


