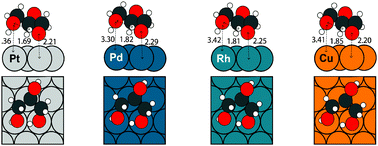
We describe an accelerated density functional theory (DFT)-based computational strategy to determine trends in the decomposition of glycerol via elementary dehydrogenation, C–C, and C–O bond scission reactions on close-packed transition metal surfaces. Beginning with periodic DFT calculations on Pt(111), the thermochemistry of glycerol dehydrogenation on Pd(111), Rh(111), Cu(111) and Ni(111) is determined using a parameter-free, bond order-based scaling relationship. By combining the results with Brønsted–Evans–Polanyi (BEP) relationships to estimate elementary reaction barriers, free energy diagrams are developed on the respective metal surfaces, and trends concerning the relative selectivity and activity for C–C and C–O bond scission in glycerol on the various metals are obtained. The results are consistent with available theoretical and experimental literature and demonstrate that scaling relationships are capable of providing powerful insights into the catalytic chemistry of complex biomolecules.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


