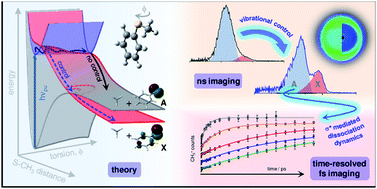
Non-adiabatic dynamics involving 1πσ* or 1nσ* excited electronic states play a key role in the photochemistry of numerous heteroatom containing aromatic (bio-)molecules. In this contribution, we investigate more exotic phenomena involved in σ* mediated dynamics, namely: (i) the role of purely quantum mechanical behavior; and (ii) manipulating non-adiabatic photochemistry through conical intersections (CIs) with ‘vibration-specific control’. This is achieved by investigating S–CH3 bond fission via a 1nσ* potential energy surface (PES) in thioanisole (C6H5SCH3). Using a combination of time- and frequency-resolved velocity map ion imaging techniques, together with ab initio calculations, we demonstrate that excitation to the 1ππ* ← S0 origin [1ππ*(ν = 0)] results in S–CH3 bond fission on the 1nσ* PES, despite an (apparent) energetic barrier to dissociation formed by a CI between the 1ππ* and 1nσ* PESs. This process occurs by accessing ‘classically forbidden’ regions of the excited state potential energy landscape where the barrier to dissociation becomes negligible, aided by torsional motion of the S–CH3 group out of the plane of the phenyl ring. Control over these dynamics is attained by populating a single quantum of the S–CH3 stretch mode in the 1ππ* state [1ππ*(ν7a = 1)], which mirrors the nuclear motion required to promote coupling through the 1ππ*/1nσ* CI, resulting in a marked change in the electronic branching in the C6H5S radical products. This observation offers an elegant contribution towards a vision of ‘quantum control’ in photo-initiated chemical reaction dynamics.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


