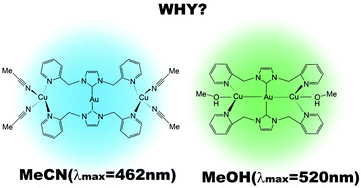
Vapochromic complexes [Au(im(CH2py)2)2(Cu(MeCN)2)2]3+1, [Au(im(CH2py)2)2(Cu(MeOH))2]3+2 and [Au(im(CH2py)2)2(Cu(H2O))2]3+3 were theoretically investigated. The Au–Cu distances of 1 and 2 (4.631 Å and 2.767 Å, respectively) optimized by the SCS-MP2 method in this work agree with the literature experimental values (4.591 Å and 2.792 Å). Their structural features are explained by computational results: (i) in 1, two MeCN molecules coordinate with the Cu center, because of the strong coordination ability of MeCN, to afford a four-coordinate tetrahedral-like Cu center. This geometry needs a long Au–Cu distance. (ii) In 2 and 3, only one MeOH or H2O molecule coordinates with the Cu center because of their weak coordination abilities, to afford a three-coordinate planar Cu center. Because the three-coordinate Cu center is flexible, the Au–Cu distance becomes short due to the Au–Cu metallophilic interaction, the strength of which is 5.3 kcal mol−1 at the SCS-MP2 level. The emission energies of 1, 2 and 3 (2.62, 2.40 and 2.38 eV, respectively) calculated here by the B3PW91 agree with their literature experimental values (2.68, 2.47, and 2.39 eV). The lowest energy triplet excited state (T1) is assigned as the excitation from the Cu d to the pyridine π* orbital in 1 and that from the Au–Cu 5d–3d anti-bonding MO to the Au–Cu 6p–4sp bonding MO in 2 and 3. As a result, the emission energy from the T1 to the ground state is different between these compounds. The difference in Au–Cu distance is one of the important factors for the differences in emission energy and assignment between 1 and others (2 and 3). The vapochromism of these compounds arises from the difference in Au–Cu distance which is determined by the balance between the strengths of the coordination of a gas molecule and the Au–Cu metallophilic interaction; in other words, the Au–Cu heterometallophilic interaction is important for the vapochromic activity of the complex.

 Please wait while we load your content...
Please wait while we load your content...

