
Ultrafast electron and energy transfer in dye-sensitized iron oxide and oxyhydroxide nanoparticles†
Abstract
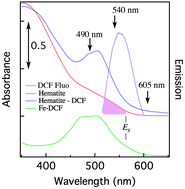
An emerging area in chemical science is the study of solid-phase redox reactions using ultrafast time-resolved spectroscopy. We have used molecules of the photoactive dye 2′,7′-dichlorofluorescein (DCF) anchored to the surface of iron(III) oxide nanoparticles to create iron(II) surface atoms via photo-initiated interfacial electron transfer. This approach enables time-resolved study of the fate and mobility of electrons within the solid phase. However, complete analysis of the ultrafast processes following dye photoexcitation of the sensitized iron(III) oxide nanoparticles has not been reported. We addressed this topic by performing femtosecond transient absorption (TA) measurements of aqueous suspensions of uncoated and DCF-sensitized iron oxide and oxyhydroxide nanoparticles, and an aqueous iron(III)–dye complex. Following light absorption, excited state relaxation times of the dye of 115–310 fs were found for all samples. Comparison between TA dynamics on uncoated and dye-sensitized hematite nanoparticles revealed the dye de-excitation pathway to consist of a competition between electron and energy transfer to the nanoparticles. We analyzed the TA data for hematite nanoparticles using a four-state model of the dye-sensitized system, finding electron and energy transfer to occur on the same ultrafast timescale. The interfacial electron transfer rates for iron oxides are very close to those previously reported for DCF-sensitized titanium dioxide (for which dye–oxide energy transfer is energetically forbidden) even though the acceptor states are different. Comparison of the alignment of the excited states of the dye and the unoccupied states of these oxides showed that the dye injects into acceptor states of different symmetry (Ti t2gvs. Fe eg).
 Please wait while we load your content...
Please wait while we load your content...

