
Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures
Abstract
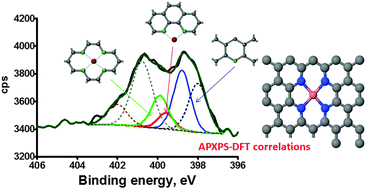
Our results validate the use of independent DFT predicted BE shifts for defect identification and constraining ambient pressure XPS observations for Me–Nx moieties in pyrolyzed carbon based ORR electrocatalysts. This supports the understanding of such
 Please wait while we load your content...
Please wait while we load your content...

