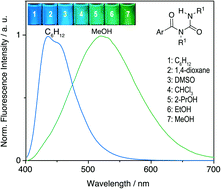
The influence of the substituent of the N-aroylurea functionality on the solvatochromic properties of this class of compounds was investigated with eight examples. The absorption spectra of these compounds exhibit the characteristic spectroscopic properties of the corresponding arene fragment and are only slightly dependent on the solvent. In contrast, all investigated aroylurea derivatives exhibit a strong solvatochromism with a good linear correlation between the emission energy and the acceptor numbers (AN) of the solvents; that is, the emission maximum shifts bathochromically (Δλ = 50–93 nm) with increasing AN. Furthermore, in media with increasing viscosity, as established in glycerol or ethanol solutions with decreasing temperature, the emission maxima are significantly shifted to shorter wavelengths and the full width at half maximum (FWHM) changes. All experimental data point to two emitting states, namely the locally excited (LE) state and the charge-transfer (CT) state. Thus, after initial photoexcitation to the LE state an internal charge transfer (ICT) takes place due to the donor–acceptor interplay between the arene unit and the N-acylureido functionality, mainly assisted by the intramolecular hydrogen bond between the terminal NH group and the aryl-substituted carbonyl functionality, hence interconverting the latter to a stronger acceptor. In the polarized CT state the acylurea unit develops a negative charge, which, after solvent relaxation, is stabilized by solvents with high acceptor number. Time-resolved emission spectroscopy revealed additional conformational changes in the excited state. Two emissive species were identified at room temperature, whose lifetimes depend strongly on the chemical environment. In addition, time-resolved emission spectra (TRES) showed red-shifted emission bands at longer delays after the excitation pulse in polar solvents. These findings are rationalized by the presence of two different emitting rotational conformers.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
