
Conformational mapping and energetics of saccharide–aromatic residue interactions: implications for the discrimination of anomers and epimers and in protein engineering†
Abstract
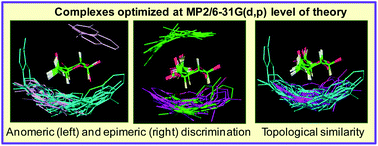
Aromatic residues play a key role in saccharide-binding sites. Experimental studies have given an estimate of the energetics of saccharide–aromatic residue interactions. In this study, dependence of the energetics on the mutual position-orientation (PO) of saccharide and aromatic residue has been investigated by geometry optimization of a very large number (164) of complexes at MP2/6-31G(d,p) level of theory. The complexes are of Tyr and Phe analogs with α/β-D-Glc, β-D-Gal, α-D-Man and α/β-L-Fuc. A number of iso-energy POs are found for the complexes of all six saccharides. Stacking and non-stacking modes of binding are found to be of comparable strengths. In general, complexes of p-OHTol are stronger than those of Tol, and those dominated by OH⋯O interactions are more stable than ones dominated by CH⋯π interactions. The strengths of OH⋯O/π interactions, but not those of CH⋯π, show large variations. Even though an aromatic residue has a large variety of POs to interact with a saccharide, distinct preferences are found due to anomeric and epimeric differences. An aromatic residue can interact from either the a- or b-face of Glc, but only through the b-face with Gal, its C4-epimer. In contrast, stacking interaction with Man (C2-epimer of Glc) requires the participation of the –CH2OH group and free rotation of this group, as is observed in solution, precludes all modes of stacking interactions. It is also found that an aromatic residue can be strategically placed either to discriminate or to accommodate (i) anomers of Glc and of Fuc and (ii) Gal/Fuc. Thus, analysis of the optimized geometries of by far the largest number of complexes, and with six different saccharides, at this level of theory has given insights into how Nature cleverly uses aromatic residues to fine tune saccharide specificities of
 Please wait while we load your content...
Please wait while we load your content...

