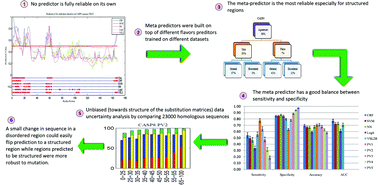
A grand challenge in the proteomics and structural genomics era is the prediction of protein structure, including identification of those proteins that are partially or wholly unstructured. A number of predictors for identification of intrinsically disordered proteins (IDPs) have been developed over the last decade, but none can be taken as a fully reliable on its own. Using a single model for prediction is typically inadequate because prediction based on only the most accurate model ignores model uncertainty. In this paper, we present an empirical method to specify and measure uncertainty associated with disorder predictions. In particular, we analyze the uncertainty in the reference model itself and the uncertainty in data. This is achieved by training a set of models and developing several meta predictors on top of them. The best meta predictor achieved comparable or better results than any other single model, suggesting that incorporating different aspects of protein disorder prediction is important for the disorder prediction task. In addition, the best meta-predictor had more balanced sensitivity and specificity than any individual model. We also assessed the effects of changes in disorder prediction as a function of changes in the protein sequence. For collections of homologous sequences, we found that mutations caused many of the predicted disordered residues to be flipped to be predicted as ordered residues, while the reverse was observed much less frequently. These results suggest that disorder tendencies are more sensitive to allowed mutations than structure tendencies and the conservation of disorder is indeed less stable than conservation of structure. Availability: five meta-predictors and four single models developed for this study will be publicly freely accessible for non-commercial use.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
