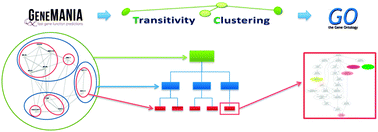
High-throughput biological experiments commonly result in a list of genes or proteins of interest. In order to understand the observed changes of the genes and to generate new hypotheses, one needs to understand the functions and roles of the genes and how those functions relate to the experimental conditions. Typically, statistical tests are performed in order to detect enriched Gene Ontology categories or pathways, i.e. the categories are observed in the genes of interest more often than is expected by chance. Depending on the number of genes and the complexity and quantity of functions in which they are involved, such an analysis can easily result in hundreds of enriched terms. To this end we developed DEFOG, a web-based application that facilitates the functional analysis of gene sets by hierarchically organizing the genes into functionally related modules. Our computational pipeline utilizes three powerful tools to achieve this goal: (1) GeneMANIA creates a functional consensus network of the genes of interest based on gene-list-specific data fusion of hundreds of genomic networks from publicly available sources; (2) Transitivity Clustering organizes those genes into a clear hierarchy of functionally related groups, and (3) Ontologizer performs a Gene Ontology enrichment analysis on the resulting gene clusters. DEFOG integrates this computational pipeline within an easy-to-use web interface, thus allowing for a novel visual analysis of gene sets that aids in the discovery of potentially important biological mechanisms and facilitates the creation of new hypotheses. DEFOG is available at http://www.mooneygroup.org/defog.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
