
Perceiving molecular themes in the structures and bonding of intermetallic phases: the role of Hückel theory in an ab initio era†‡
Abstract
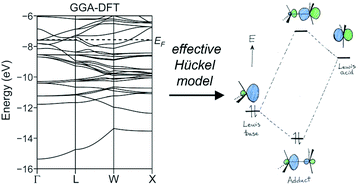
Qualitative molecular orbital theory is central to our understanding of the bonding and reactivity of molecules and materials across chemistry. Advances in computational technology and methodology, however, have made ab initio or density functional theory calculations a simpler alternative, offering reliable results on increasingly large systems in a reasonable time-scale without the need for concerns about the approximations and parameterization of semi-empirical one-electron based methods. In this perspective, we illustrate how the availability of higher-level computational results can augment, rather than supplant, the insights provided by approaches such as the simple and extended Hückel methods. We begin by describing a way to parameterize Hückel-type Hamiltonians against DFT results for intermetallic systems. The potential for chemical understanding embodied by such orbital-based models is then demonstrated with two schemes of bonding analysis that originated in them (but can be extended to DFT results): the μ3-acid/base model and the μ2-Hückel chemical pressure analysis, which translate the molecular concepts of acidity and electronic/steric competition, respectively, into the context of intermetallic chemistry.
- This article is part of the themed collection: New Talent: Americas
 Please wait while we load your content...
Please wait while we load your content...

