
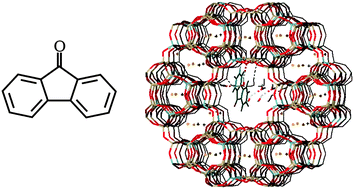
First-principles simulation of the absorption bands of fluorenone in zeolite L†
Abstract
The ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C motif is retained in all conformations. Although the interactions between K+ and the fluorenone carbonyl group result in an average lengthening of the CO bond and in a redshift of the lowest energy absorption band compared to gas phase or
C motif is retained in all conformations. Although the interactions between K+ and the fluorenone carbonyl group result in an average lengthening of the CO bond and in a redshift of the lowest energy absorption band compared to gas phase or
 Please wait while we load your content...
Please wait while we load your content...

