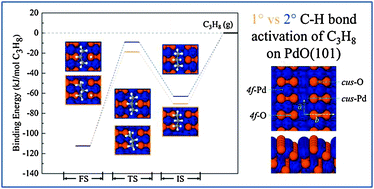
We used dispersion-corrected density functional theory (DFT-D3) calculations to investigate the initial C–H bond cleavage of propane σ-complexes adsorbed on the PdO(101) surface. The calculations predict that propane molecules adsorbed in η1 configurations can undergo facile C–H bond cleavage on PdO(101), where the energy barrier for C–H bond activation is lower than that for desorption for each molecular complex. The preferred pathway for propane dissociation on PdO(101) corresponds to cleavage of a primary C–H bond of a so-called staggered p-2η1 complex which initially coordinates with the surface by forming two H–Pd dative bonds, one at each CH3 group. Among all of the adsorbed propane complexes, the staggered p-2η1 complex has the highest binding energy and must overcome the lowest energy barrier for C–H bond scission. Analysis of the atomic charges reveals that propane C–H bond cleavage occurs heterolytically on PdO(101), and suggests that primary C–H bond activation is favored because a more stabilizing charge distribution develops within the 1-propyl transition state structures. Lastly, we conducted kinetic simulations using microkinetic models derived from the DFT-D3 structures, and find that the models reproduce the apparent activation energy for propane dissociation on PdO(101) to within 14% of that determined experimentally. We show that the entropic contributions of the adsorbed transition structures greatly exceed those predicted by the harmonic oscillator model, and that quantitative agreement with the apparent dissociation pre-factor may be obtained by approximating two of the frustrated adsorbate motions as free motions while treating the remaining modes as harmonic vibrations.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


