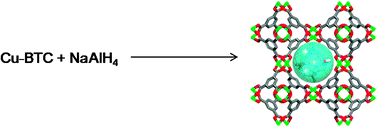
Reactive nanoparticles are of great interest for applications ranging from catalysis to energy storage. However, efforts to relate cluster size to thermodynamic stability and chemical reactivity are hampered by broad pore size distributions and poorly characterized chemical environments in many microporous templates. Metal hydrides are an important example of this problem. Theoretical calculations suggest that reducing their critical dimension to the nanoscale can in some cases considerably destabilize these materials and there is clear experimental evidence for accelerated kinetics, making hydrogen storage applications more attractive in some cases. However, quantitative measurements establishing the influence of size on thermodynamics are lacking, primarily because carbon aerogels often used as supports provide inadequate control over size and pore chemistry. Here, we employ the nanoporous metal–organic framework (MOF) Cu-BTC (also known as HKUST-1) as a template to synthesize and confine the complex hydride NaAlH4. The well-defined crystalline structure and monodisperse pore dimensions of this MOF allow detailed, quantitative probing of the thermodynamics and kinetics of H2 desorption from 1-nm NaAlH4 clusters (NaAlH4@Cu-BTC) without the ambiguity associated with amorphous templates. Hydrogen evolution rates were measured as a function of time and temperature using the Simultaneous Thermogravimetric Modulated Beam Mass Spectrometry method, in which sample mass changes are correlated with a complete analysis of evolved gases. NaAlH4@Cu-BTC undergoes a single-step dehydrogenation reaction in which the Na3AlH6 intermediate formed during decomposition of the bulk hydride is not observed. Comparison of the thermodynamically controlled quasi-equilibrium reaction pathways in the bulk and nanoscale materials shows that the nanoclusters are slightly stabilized by confinement, having an H2 desorption enthalpy that is 7 kJ (mol H2)−1 higher than the bulk material. In addition, the activation energy for desorption is only 53 kJ (mol H2)−1, more than 60 kJ (mol H2)−1 lower than the bulk. When combined with first-principles calculations of cluster thermodynamics, these data suggest that although interactions with the pore walls play a role in stabilizing these particles, size exerts the greater influence on the thermodynamics and reaction rates.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


