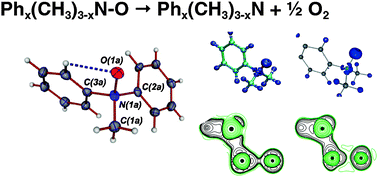
The bonding situation and energetics of the N–O bond in a series of amine-N-oxides, Phx(CH3)3−xN–O, where x = 0–3, were analyzed experimentally and theoretically. There is a notable nearly linear decrease of the N–O bond dissociation energies (BDEs) for this series with an increasing number of phenyl groupsx. This was investigated experimentally by X-ray high angle multipole refinement techniques in combination with subsequent topological analysis of the electron density for the representative (CH3)2PhN–O, 2, and complementary theoretical calculations at the DFT and multireference CASSCF and MR-perturbation theory (MCQDPT2) levels. Both the theoretical and experimental results unambiguously revealed a polar covalent σ-bond for the N–O bond with an essentially identical bonding situation for all amine-N-oxides studied. This apparent disparity between the bonding situation and the trend of BDEs is attributed to the large differences of the relaxation energies of the corresponding amines Phx(CH3)3−xN, (x = 0–3), respectively, the required preparation energies (ΔEprep) for the reverse N–O bond forming process. The detailed theoretical analysis of the amines allowed us to trace the trend of larger values of ΔEprep for a higher number of phenyl groupsx to an increase of n(N) → π*(C–C) delocalization interactions.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


