
Prediction on the existence and chemical stability of cuprous fluoride
Abstract
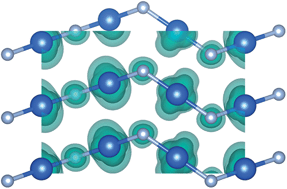
The existence of CuF has been a matter of debate for the past century. A 1933 report of the synthesis of CuF in the sphalerite structure has never been reproduced, however, it consistently appears in textbooks and databases. We report the results from a computational study of CuF based on a hybrid density functional theory (DFT) approach and identify the cinnabar crystal structure as an energy minimum, which incorporates linear F–Cu–F chains that are characteristic of the Cu(I) ion. Assessment of the oxidation and disproportionation reactions reveals that while CuF is thermodynamically stable with respect to the standard state, it can be oxidised readily to form CuF2. Moreover, ab initio molecular dynamics simulations reveal that the linear F–Cu–F chains have a low barrier to rotation, so that at moderate temperatures the material might not give rise to a clear diffraction pattern. The predicted ionization potential of 6.5 eV, with respect to the vacuum level, suggests that the material may be suitable for photochemical applications through the formation of a heterostructure with Cu2O and/or ZnO.
 Please wait while we load your content...
Please wait while we load your content...

