
Long-timescale simulations of diffusion in molecular solids
Abstract
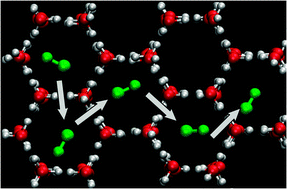
Kinetic processes play a crucial role in the formation and evolution of molecular layers. In this perspective we argue that adaptive kinetic Monte Carlo is a powerful simulation technique for determining key kinetic processes in molecular solids. The applicability of the method is demonstrated by simulating the diffusion of a CO admolecule on a water ice surface, which is an important process for the formation of organic compounds on interstellar dust grains. CO diffusion is found to follow Arrhenius behavior and the corresponding effective
 Please wait while we load your content...
Please wait while we load your content...

