
Comparison of explicitly correlated local coupled-cluster methods with various choices of virtual orbitals†
Abstract
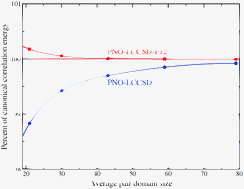
Explicitly correlated local coupled-cluster (LCCSD-F12) methods with pair natural orbitals (PNOs), orbital specific virtual orbitals (OSVs), and projected atomic orbitals (PAOs) are compared. In all cases pair-specific virtual subspaces (domains) are used, and the convergence of the correlation energy as a function of the domain sizes is studied. Furthermore, the performance of the methods for reaction energies of 52 reactions involving 58 small and medium sized molecules is investigated. It is demonstrated that for all choices of virtual orbitals much smaller domains are needed in the explicitly correlated methods than without the explicitly correlated terms, since the latter correct a large part of the domain error, as found previously. For PNO-LCCSD-F12 with VTZ-F12 basis sets on the average only 20 PNOs per pair are needed to obtain reaction energies with a root mean square deviation of less than 1 kJ mol−1 from complete basis set estimates. With OSVs or PAOs at least 4 times larger domains are needed for the same accuracy. A new hybrid method that combines the advantages of the OSV and PNO methods is proposed and tested. While in the current work the different local methods are only simulated using a conventional CCSD program, the implications for low-order scaling local implementations of the various methods are discussed.
- This article is part of the themed collection: Fragment and localized orbital methods in electronic structure theory
 Please wait while we load your content...
Please wait while we load your content...

