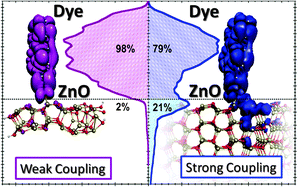
We report a first principles density functional theory/time-dependent density functional theory (DFT/TDDFT) computational investigation on a prototypical perylene dye anchored to realistic ZnO nanostructures, approaching the size of the ZnO nanowires used in dye-sensitized solar cells devices. DFT calculations were performed on (ZnO)n clusters of increasing size, with n up to 222, of 1.3 × 1.5 × 3.4 nm dimensions, and for the related dye-sensitized models. We show that quantum confinement in the ZnO nanostructures substantially affects the dye/semiconductor alignment of energy levels, with smaller ZnO models providing unfavourable electron injection. An increasing broadening of the dye LUMO is found moving to larger substrates, substantially contributing to the interfacial electronic coupling. TDDFT excited state calculations for the investigated dye@(ZnO)222 system are fully consistent with experimental data, quantitatively reproducing the red-shift and broadening of the visible absorption spectrum observed for the ZnO-anchored dye compared to the dye in solution. TDDFT calculations on the fully interacting system also introduce a contribution to the dye/semiconductor admixture, due to configurational excited state mixing. Our results highlight the importance of quantum confinement in dye-sensitized ZnO interfaces, and provide the fundamental insight lying at the heart of the associated DSC devices.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


