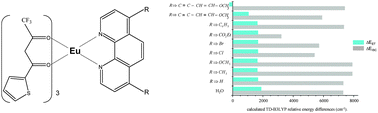
Density functional theory/time-dependent density functional theory (DFT/TD-DFT) calculations were performed on a series of europium(III) complexes of 4,7-disubstituted-1,10-phenanthroline ligands (phen-X) of general formula Eu(TTA)3(phen-X) (where TTA stands for thenoyltrifluoroacetonato and X = H, CH3, OCH3, Cl, Br, CO2Et, C6H5, C4HOCH3 and C4H3OCH3). The effect of such substitution on the structural, electronic and photophysical properties is established. Absorption spectra calculations show that different phen substituents have different effects on absorption peak positions and on transition characters while only substituent's influence via extended π-conjugation of the phen ligand can effectively tune the triplet state. Considering the ΔEISC and ΔEET values, the luminescent 5D0 state of the Eu3+ ion can be efficiently populated in most complexes. The exceptions are the complexes with CO2Et and C4H3OCH3 groups.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?