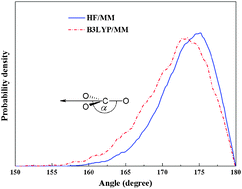
Two combined quantum mechanics/molecular mechanics (QM/MM) molecular dynamics simulations, namely HF/MM and B3LYP/MM, have been performed to investigate the local hydration structure and dynamics of carbonate (CO32−) in dilute aqueous solution. With respect to the QM/MM scheme, the QM region, which contains the CO32− and its surrounding water molecules, was treated at HF and B3LYP levels of accuracy, respectively, using the DZV+ basis set, while the rest of the system is described by classical MM potentials. For both the HF/MM and B3LYP/MM simulations, it is observed that the hydrogen bonds between CO32− oxygens and their nearest-neighbor waters are relatively strong, i.e., compared to water–water hydrogen bonds in the bulk, and that the first shell of each CO32−oxygen atom somewhat overlaps with the others, which allows migration of water molecules among the coordinating sites to exist. In addition, it is observed that first-shell waters are either “loosely” or “tightly” bound to the respective CO32−oxygen atoms, leading to large fluctuations in the number of first-shell waters, ranging from 1 to 6 (HF/MM) and 2 to 7 (B3LYP/MM), with the prevalent value of 3. Upon comparing the HF and B3LYP methods in describing this hydrated ion, the latter is found to overestimate the hydrogen-bond strength in the CO32−–water complexes, resulting in a slightly more compact hydration structure at each of the CO32− oxygens.

 Please wait while we load your content...
Please wait while we load your content...

