
Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations
Abstract
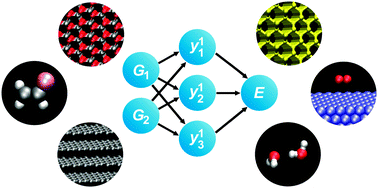
The accuracy of the results obtained in molecular dynamics or Monte Carlo simulations crucially depends on a reliable description of the atomic interactions. A large variety of efficient potentials has been proposed in the literature, but often the optimum functional form is difficult to find and strongly depends on the particular system. In recent years, artificial neural networks (NN) have become a promising new method to
 Please wait while we load your content...
Please wait while we load your content...

