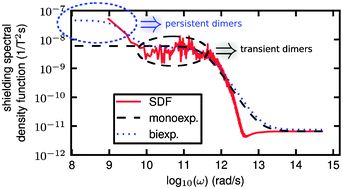
Nuclear spin relaxation provides detailed dynamical information on molecular systems and materials. Here, first-principles modeling of the chemical shift anisotropy (CSA) relaxation time for the prototypic monoatomic 129Xe gas is carried out, both complementing and predicting the results of NMR measurements. Our approach is based on molecular dynamics simulations combined with pre-parametrized ab initio binary nuclear shielding tensors, an “NMR force field”. By using the Redfield relaxation formalism, the simulated CSA time correlation functions lead to spectral density functions that, for the first time, quantitatively determine the experimental spin–lattice relaxation times T1. The quality requirements on both the Xe–Xe interaction potential and binary shielding tensor are investigated in the context of CSA T1. Persistent dimers Xe2 are found to be responsible for the CSA relaxation mechanism in the low-density limit of the gas, completely in line with the earlier experimental findings.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


