
Computing the 7Li NMR chemical shielding of hydrated Li+ using cluster calculations and time-averaged configurations from ab initio molecular dynamics simulations†
Abstract
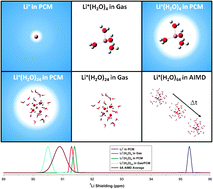
Ab initio molecular dynamics (AIMD) simulations have been used to predict the time-averaged Li
 Please wait while we load your content...
Please wait while we load your content...

