
Quantifying the breakdown of the Born–Oppenheimer approximation in surface chemistry
Abstract
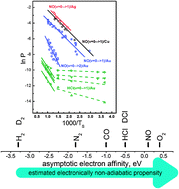
The Born–Oppenheimer Approximation (BOA) forms the basis for calculating electronically adiabatic potential energy surfaces, thus providing the framework for developing a molecular level understanding of a variety of important chemical problems. For surface chemistry at metal surfaces, it is now clear that for some processes electronically nonadiabatic effects can be important, even dominant; however, the magnitude of BOA breakdown may vary widely from one chemical system to another. In this paper we show that molecular-beam surface scattering experiments can be used to derive quantitative information about the magnitude of BOA breakdown. A state-to-state rate model is used to interpret the pre-exponential factor of the well-known Arrhenius surface temperature dependence of the electronically nonadiabatic vibrational excitation. We also show that reference to a “thermal limit” provides a quick and simple rule of thumb for quantifying BOA breakdown. We demonstrate this approach by comparing electronically nonadiabatic vibrational inelasticity for NO(ν = 0 → 1) to NO(ν = 15 → ν′ ≪ 15) and show that the electronically nonadiabatic coupling strengths are of a similar magnitude. We compare experiments for NO and HCl scattering from Au(111) and derive the quantitative relative magnitude for the electronically nonadiabatic influences in each system. The electronically nonadiabatic influences are 300–400 times larger for NO than for HCl, for incidence energies near 0.9 eV.
 Please wait while we load your content...
Please wait while we load your content...

