
A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions†
Abstract
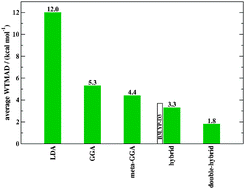
A thorough energy benchmark study of various density functionals (DFs) is carried out with the new GMTKN30 database for general main group thermochemistry, kinetics and noncovalent interactions [Goerigk and Grimme, J. Chem. Theor. Comput., 2010, 6, 107; Goerigk and Grimme, J. Chem. Theor. Comput., 2011, 7, 291]. In total, 47 DFs are investigated: two LDAs, 14 GGAs, three meta-GGAs, 23 hybrids and five double-hybrids. Besides the double-hybrids, also other modern approaches, i.e., the M05 and M06 classes of functionals and range-separated hybrids, are tested. For almost all functionals, the new DFT-D3 correction is applied in order to consistently test the performance also for important noncovalent interactions; the parameters are taken from previous works or determined for the present study. Basis set and quadrature grid issues are also considered. The general aim of the study is to work out which functionals are generally well applicable and robust to describe the energies of molecules. In summary, we recommend on the GGA level the B97-D3 and revPBE-D3 functionals. The best meta-GGA is oTPSS-D3 although meta-GGAs represent in general no clear improvement compared to numerically simpler GGAs. Notably, the widely used B3LYP functional performs worse than the average of all tested hybrids and is also very sensitive to the application of dispersion corrections. We discourage its usage as a standard method without closer inspection of the results, as it still seems to be often done nowadays. Surprisingly, long-range corrected exchange functionals do in general not perform better than the corresponding standard hybrids. However, the ωB97X-D functional seems to be a promising method. The most robust hybrid is Zhao and Truhlar's PW6B95 functional in combination with DFT-D3. If higher accuracy is required, double-hybrids should be applied. The corresponding DSD-BLYP-D3 and PWPB95-D3 variants are the most accurate and robust functionals of the entire study. Additional calculations with MP2 and and its spin-scaled variants SCS-MP2, S2-MP2 and SOS-MP2 revealed that double-hybrids in general outperform those. Only SCS-MP2 can be recommended, particularly for reaction energies. We suggest its usage when a large self-interaction error is expected that prohibits usage of double-hybrids. Perdews' metaphoric picture of Jacob's Ladder for the classification of density functionals' performance could unbiasedly be confirmed with GMTKN30. We also show that there is no statistical correlation between a functional's accuracy for
 Please wait while we load your content...
Please wait while we load your content...

