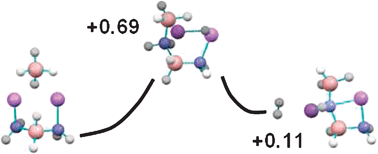
Electronic structure calculations have been used to determine and compare the thermodynamics of H2 release from ammonia borane (NH3BH3), lithium amidoborane (LiNH2BH3), and sodium amidoborane (NaNH2BH3). Using two types of exchange correlation functional we show that in the gas-phase the metal amidoboranes have much higher energies of complexation than ammonia borane, meaning that for the former compounds the B–N bond does not break upon dehydrogenation. Thermodynamically however, both the binding energy for H2 release and the activation energy for dehydrogenation are much lower for NH3BH3 than for the metal amidoboranes, in contrast to experimental results. We reconcile this by also investigating the effects of dimer complexation (2×NH3BH3, 2×LiNH2BH3) on the dehydrogenation properties. As previously described in the literature the minimum energy pathway for H2 release from the 2×NH3BH3 complex involves the formation of a diammoniate of diborane complex ([BH4]−[NH3BH2NH3]+). A new mechanism is found for dehydrogenation from the 2×LiNH2BH3 dimer that involves the formation of an analogous dibroane complex ([BH4]−[LiNH2BH2LiNH2]+), intriguingly it is lower in energy than the original dimer (by 0.13 eV at ambient temperatures). Additionally, this pathway allows almost thermoneutral release of H2 from the lithium amidoboranes at room temperature, and has an activation barrier that is lower in energy than for ammonia borane, in contrast to other theoretical research. The transition state for single and dimer lithium amidoborane demonstrates that the light metal atom plays a significant role in acting as a carrier for hydrogen transport during the dehydrogenation process via the formation of a Li–H complex. We posit that it is this mechanism which is responsible, in condensed molecular systems, for the improved dehydrogenation thermodynamics of metal amidoboranes.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


