
Are ab initio quantum chemistry methods able to predict vibrational states up to the dissociation limit for multi-electron molecules close to spectroscopic accuracy?†
Abstract
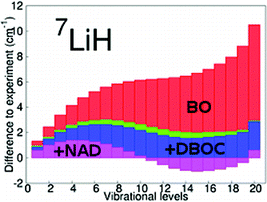
The aim of the study was to explore the limits of ab initio methods towards the description of excited vibrational levels up to the dissociation limit for molecules having more than two electrons. To this end a high level ab initio potential energy function was constructed for the four-electron LiH molecule in order to accurately predict a complete set of bound vibrational levels corresponding to the electronic ground state. It was composed from: (a) an ab initio non-relativistic potential obtained at the MR-CISD level including size-extensivity corrections and quintuple-sextuple ζ extrapolation of the basis, (b) MVD relativistic corrections obtained at icMR-CISD/cc-pwCV5Z level, and (c) DBOC obtained at the MR-CISD/cc-pwCVTZ level. Finally, the importance of non-adiabatic effects was also tested by using atomic masses in the vibrational kinetic energy operator. The calculated vibrational levels were compared with those obtained from
 Please wait while we load your content...
Please wait while we load your content...

