The synthesis of [(1,2,4-tBu3C5H2)FeI]2, [Cp′FeI]2 (1), and its reaction chemistry are described. Complex 1 shows remarkable stability toward ligand redistribution in solution and gas-phase due to kinetic stabilization with respect to 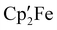 formation. In coordinating solvents the dimer is homolytically cleaved to form paramagnetic mono-solvate 16 valence-electron (VE) adducts [Cp′FeI(L)]. These solvent adducts are labile, and 1 is re-established on exposure to dynamic vacuum. Complex 1 was demonstrated to be a suitable synthon for [Cp′Fe]+ transfer to Ir pincer complexes and successful η6-coordination of this fragment to the arene ring of κ1-metallated POCOP–iridium pincer complexes [(Cp′Fe)(POCOP)Ir(H)(I)] ([5]++) and [(Cp′Fe)(POCOP)Ir(C2H4)] ([7]++) (POCOP = C6H3-2,6-(OPtBu2)2) was accomplished. In these heterometallic organometallic complexes, the π- and σ-electrons of the POCOP-pincer phenyl anion are involved in η6- and κ1-coordination to iron(II) and iridium(III) and iridium(I), respectively. [Cp′Fe]+ coordination alters the charge distribution in the (POCOP)Ir complex by inducing significant charge transfer from the POCOP aryl π-system to the [Cp′Fe]+ fragment, and therefore yielding an electron-poor and more electrophilic iridium center. Density functional theory (DFT) calculations provide a detailed understanding of this effect. Preliminary catalytic studies show that [7]++ is an active catalyst in transfer dehydrogenation between cyclooctane (COA) and tert-butyl ethylene (TBE).
formation. In coordinating solvents the dimer is homolytically cleaved to form paramagnetic mono-solvate 16 valence-electron (VE) adducts [Cp′FeI(L)]. These solvent adducts are labile, and 1 is re-established on exposure to dynamic vacuum. Complex 1 was demonstrated to be a suitable synthon for [Cp′Fe]+ transfer to Ir pincer complexes and successful η6-coordination of this fragment to the arene ring of κ1-metallated POCOP–iridium pincer complexes [(Cp′Fe)(POCOP)Ir(H)(I)] ([5]++) and [(Cp′Fe)(POCOP)Ir(C2H4)] ([7]++) (POCOP = C6H3-2,6-(OPtBu2)2) was accomplished. In these heterometallic organometallic complexes, the π- and σ-electrons of the POCOP-pincer phenyl anion are involved in η6- and κ1-coordination to iron(II) and iridium(III) and iridium(I), respectively. [Cp′Fe]+ coordination alters the charge distribution in the (POCOP)Ir complex by inducing significant charge transfer from the POCOP aryl π-system to the [Cp′Fe]+ fragment, and therefore yielding an electron-poor and more electrophilic iridium center. Density functional theory (DFT) calculations provide a detailed understanding of this effect. Preliminary catalytic studies show that [7]++ is an active catalyst in transfer dehydrogenation between cyclooctane (COA) and tert-butyl ethylene (TBE).
![Graphical abstract: [Cp′FeI]2 as convenient entry into iron-modified pincer complexes: bimetallic η6,κ1-POCOP-pincer iron iridium compounds](/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C1NJ20399A&imageInfo.ImageIdentifier.Year=2011)
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


