
Understanding the p-type defect chemistry of CuCrO2
Abstract
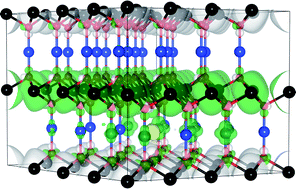
CuCrO2 is the most promising Cu-based delafossite for p-type optoelectronic devices. Despite this, little is known about the p-type conduction mechanism of this material, with both CuI/CuII and CrIII/CrIV hole mechanisms being proposed. In this article we examine the electronic structure, thermodynamic stability and the p-type defect chemistry of this ternary compound using density functional theory with three different approaches to the exchange and correlation; the generalized-gradient-approximation of Perdew, Burke and Ernzerhof (PBE), PBE with an additional correction for on-site Coulombic interactions (PBE + U) and the nonlocal, screened-exchange hybrid functional HSE06. The fundamental band gap of CuCrO2 is demonstrated to be indirect in nature. Under all growth conditions, the dominant intrinsic p-type defect will be the Cu vacancy, with hole formation centered solely on the Cu sublattice. Mg doping is found to be significantly lower in energy than intrinsic defect formation, explaining the large increases in conductivity seen experimentally. Cu-rich/Cr-poor growth conditions are found to be optimal for both intrinsic and extrinsic (Mg doping) defect formation, and should be adopted to maximize performance.
 Please wait while we load your content...
Please wait while we load your content...