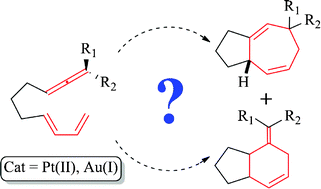
The intramolecular [4C+3C] cycloaddition reaction of allenedienes catalysed by PtCl2 and several Au(I) complexes has been studied by means of DFT calculations. Overall, the reaction mechanism comprises three main steps: (i) the formation of a metal allyl cation intermediate, (ii) a [4C(4π)+3C(2π)] cycloaddition that produces a seven-membered ring and (iii) a 1,2-hydrogen migration process on these intermediates. The reaction proceeds with complete diastereochemical control resulting from a favoured exo-like cycloaddition. Allene substituents have a critical influence in the reaction outcome and mechanism. The experimental observation of [4C+2C] cycloadducts in the reaction of substrates lacking substituents at the allene terminus can be explained through a mechanism involving Pt(IV)-metallacycles. With gold catalysts it is also possible to obtain [4C+2C] cycloaddition products, but only with substrates featuring terminally disubstituted allenes, and employing π-acceptor ligands at gold. However the mechanism for the formation of these adducts is completely different to that proposed with PtCl2, and consists of the formation of a metal allyl cation, subsequent [4C+3C] cycloaddition and a 1,2-alkyl shift (ring contraction). Electronic analysis indicates that the divergent pathways are mainly controlled by the electronic properties of the gold heptacyclic species (L-Au-C2), in particular, the backdonation capacity of the metal center to the unoccupied C2 (pπ-orbital) of the intermediate resulting from the [4C+3C] cycloaddition. The less backdonation, (i.e. using P(OR)3Au+ complexes), the more favoured is the 1,2-alkyl shift.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


