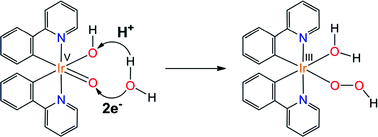
The cationic iridium complex [Ir(OH2)2(phpy)2]+ (phpy = o-phenylpyridine) is among the most efficient mononuclear catalysts for water oxidation. The postulated active species is the oxo complex [Ir(O)(X)(phpy)2]n, with X = OH2 (n = +1), OH− (n = 0) or O2− (n = −1), depending on the pH. The reactivity of these species has been studied computationally at the DFT(B3LYP) level. The three [Ir(O)(X)(phpy)2]n complexes have an electrophilic Ir(V)-oxo moiety, which yields an O–O bond by undergoing a nucleophilic attack of water in the critical step of the mechanism. In this step, water transfers one proton to either the Ir(V)-oxo moiety or the ancillary X ligand. Five different reaction pathways associated with this acid/base mechanism have been characterized. The calculations show that the proton is preferably accepted by the X ligand, which plays a key role in the reaction. The higher the basicity of X, the lower the energy barrier associated with O–O bond formation. The anionic species, [Ir(O)2(phpy)2]−, which has the less electrophilic Ir(V)-oxo moiety but the most basic X ligand, promotes O–O bond formation through the lowest energy barrier, 14.5 kcal mol−1. The other two active species, [Ir(O)(OH)(phpy)2] and [Ir(O)(OH2)(phpy)2]+, which have more electrophilic Ir(V)-oxo moieties but less basic X ligands, involve higher energy barriers, 20.2 kcal mol−1 and 25.9 kcal mol−1, respectively. These results are in good agreement with experiments showing important pH effects in similar catalytic systems. The theoretical insight given by the present study can be useful in the design of more efficient water oxidation catalysts. The catalytic activity may increase by using ligand scaffolds bearing internal bases.

 Please wait while we load your content...
Please wait while we load your content...

