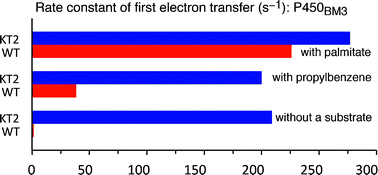
The substrate-free crystal structure of a five-mutation directed evolution variant of CYP102A1 (P450BM3) with generic activity-enhancing properties (“KT2”) has been determined to 1.9-Å resolution. There is a close resemblance to substrate-bound structures of the wild-type enzyme (WT). The disruption of two salt bridges that link the G- and I-helices in WT causes conformational changes that break several hydrogen bonds and reduce the angle of the kink in the I-helix where dioxygen activation is thought to take place. The side-chain of a key active site residue, Phe87, is rotated in one molecule of the asymmetric unit, and the side-chains of Phe158 and Phe261 cascade into the orientations found in fatty-acid-bound forms of the enzyme. The iron is out of the porphyrin plane, towards the proximal cysteine. Unusually, the axial water ligand to the haem iron is not hydrogen-bonded to Ala264. The first electron transfer from the reductase domain to the haem domain of substrate-free KT2 is almost as fast as in palmitate-bound WT even though the reduction potential of the haem domain is only slightly more oxidising than that of substrate-free WT. However, NADPH is turned over slowly in the absence of substrate, so the catalytic cycle is gated by a step subsequent to the first electron transfer—a contrast to WT. Propylbenzene binding slightly raises the first electron transfer rate in WT but not in KT2. It is proposed that the generic rate accelerating properties of KT2 arise from the substrate-free form being in a catalytically ready conformation, such that substrate-induced changes to the structure play a less significant role in promoting the first electron transfer than in WT.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


