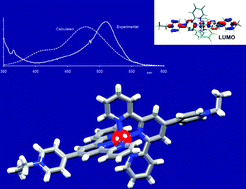
A series of N-alkylated derivatives [RuL2][PF6]4 has been prepared from [Ru(pytpy)2][PF6]2 (N-alkyl substituent = 4-cyanobenzyl, 4-nitrobenzyl, ethyl, cyanomethyl, allyl, octyl). Solution NMR spectroscopic, electrochemical and photophysical properties are reported, along with the single crystal structure of [Ru(4)2][PF6]4·H2O (4 = 4′-(4-(1-ethylpyridinio))-2,2′:6′,2′-terpyridine). Anion exchange leads to the water-soluble [RuL2][HSO4]4 salts (N-alkyl substituent = benzyl, 4-cyanobenzyl, 4-nitrobenzyl, ethyl, cyanomethyl, allyl, octyl) and the NMR spectroscopic signatures of pairs of hexafluoridophosphate and hydrogensulfate salts are compared. The change in anion has little effect on the energies of absorptions in the electronic spectra, although for all complexes, decreases in extinction coefficients are observed. The emission spectra and lifetimes for the hexafluoridophosphate and hydrogensulfate salts show similar trends; all exhibit an emission close to 720–730 nm (λex = 510 nm). For a given ligand, L, the emission lifetime decreases on going from [RuL2][PF6]4 to [RuL2][HSO4]4. However, trends are the same for both salts, i.e. the longest lived emitters are observed for N-ethyl, N-octyl and N-benzyl derivatives, and the shortest lived emitters are those containing cyano or nitro groups. Significantly, in the absorption spectra of the complexes, there is little variation in the energy of the MLCT band, suggesting that the character of the ligand orbital involved in the transition contains no character from the N-substituent. We have addressed this by carrying out a complementary DFT and TD-DFT study. Calculated absorption spectra predict a red shift in λmax on going from [Ru(pytpy)2]2+ to [RuL2]4+, and little variation in λmax within the series of [RuL2]4+ complexes; these results agree with experimental observations. Analysis of the compositions of the MOs involved in the MLCT transitions explain the experimental observations, showing that there is no contribution from orbitals on the N-alkyl substituents, consistent with the fact that the nature of the N-substituents has little influence on the energy of the MLCT band. The theoretical results also reveal satisfactory agreement between calculated and crystallographic data for [Ru(1)2]4+ (1 = 4′-(4-(1-benzylpyridinio))-2,2′:6′,2′-terpyridine) and [Ru(4)2]4+.

 Please wait while we load your content...
Please wait while we load your content...

