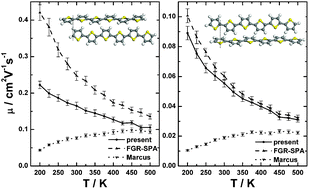
The electronic coupling between adjacent molecules is an important parameter for the charge transport properties of organic semiconductors. In a previous paper, a semiclassical generalized nonadiabatic transition state theory was used to investigate the nonperturbative effect of the electronic coupling on the charge transport properties, but it is not applicable at low temperatures due to the presence of high-frequency modes from the intramolecular conjugated carbon–carbon stretching vibrations [G. J. Nan et al., J. Chem. Phys., 2009, 130, 024704]. In the present paper, we apply a quantum charge transfer rate formula based on the imaginary-time flux–flux correlation function without the weak electronic coupling approximation. The imaginary-time flux–flux correlation function is then expressed in terms of the vibrational-mode path average and is evaluated by the path integral approach. All parameters are computed by quantum chemical approaches, and the mobility is obtained by kinetic Monte-Carlo simulation. We evaluate the intra-layer mobility of sexithiophene crystal structures in high- and low-temperature phases for a wide range of temperatures. In the case of strong coupling, the quantum charge transfer rates were found to be significantly smaller than those calculated using the weak electronic coupling approximation, which leads to reduced mobility especially at low temperatures. As a consequence, the mobility becomes less dependent on temperature when the molecular packing leads to strong electronic coupling in some charge transport directions. The temperature-independent charge mobility in organic thin-film transistors from experimental measurements may be explained from the present model with the grain boundaries considered. In addition, we point out that the widely used Marcus equation is invalid in calculating charge carrier transfer rates in sexithiophene crystals.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


